| 12 May 2022 |
|
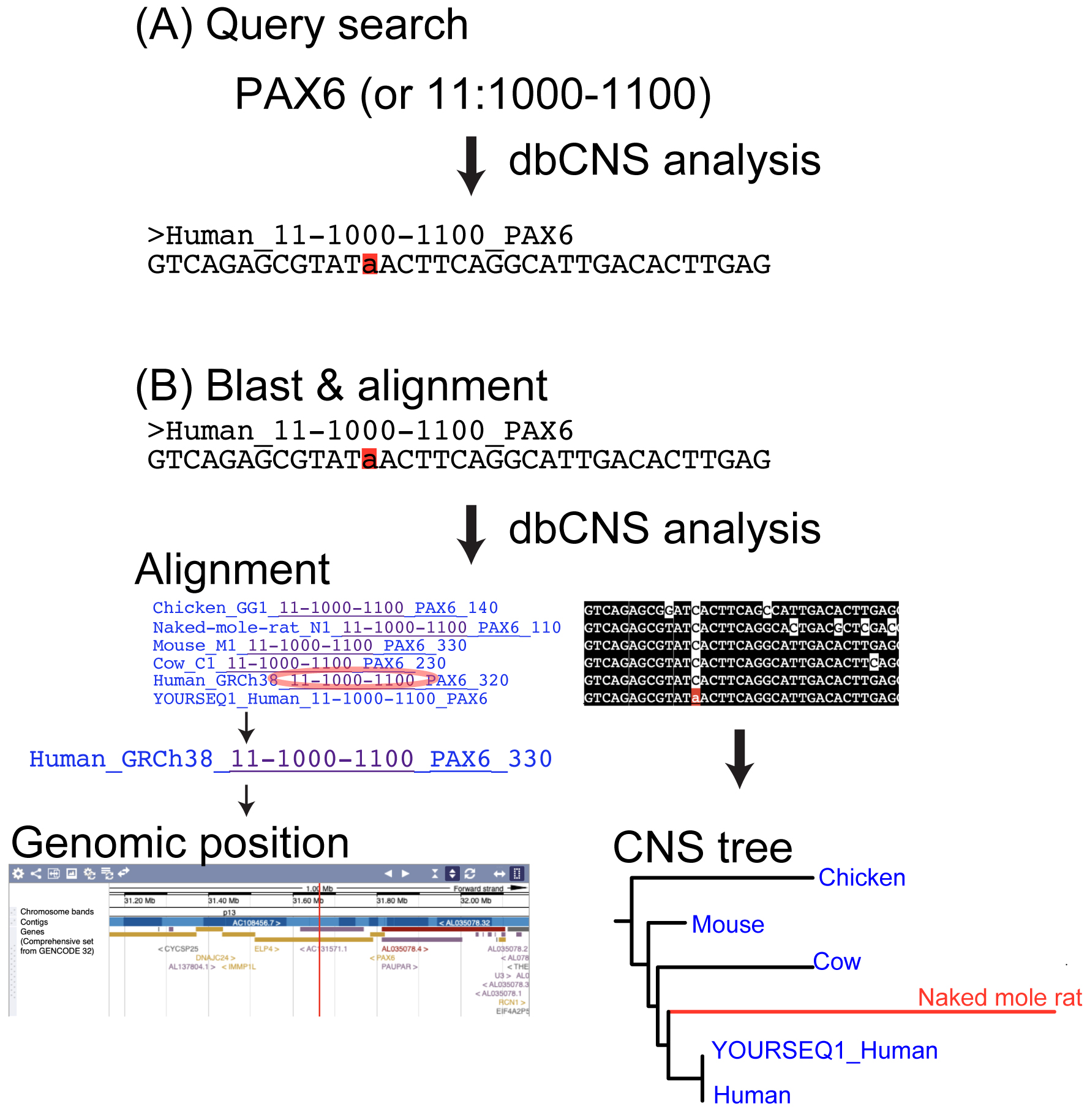
dbCNS (http://yamasati.nig.ac.jp/dbcns) is a database and an identification pipeline for conserved non-coding sequences (CNSs) of vertebrates. The database contains 2 sources: more than 6,800,000 published CNSs and 180 genome sequences.
Users can
- find CNSs near genes of interest from the database by uploading keywords or coordinates.
- construct multiple sequence alignments and CNS trees by uploading CNS sequences as queries.
|
|
|
NIG (from 27 Jan 2020)
http://yamasati.nig.ac.jp/dbcns
SAKURA internet (from 27 Jan 2020)
http://133.167.86.72/dbcns/
|
|
Inoue J. and Saitou N.
dbCNS: a new database for conserved non-coding sequences. Submitted.
|
|

|
|
- CNS database (link)
- Genome sequence database
|
|
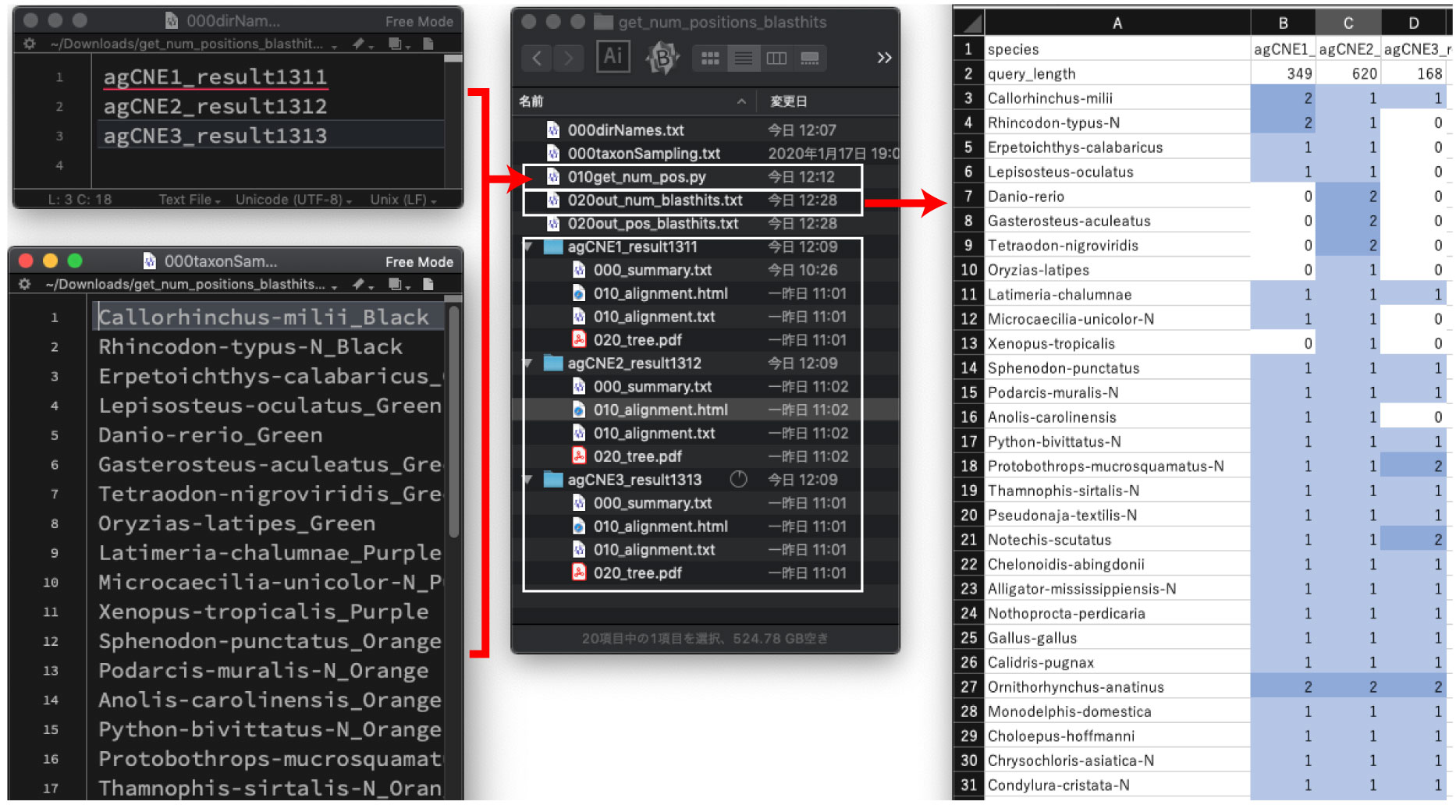
| Count the number of blast hits from multiple results |
|
In Inoue and Saitou submitted, dbCNS analyses were conducted for multiple CNS queries for gnathostome (case study 2) and teleost (case study 3) analyses. Here we show how such multiple analyses were conducted and resultant outputs were summarized.
|
| Multiple analyses |
We use 3 CNS examples:
>agCNE1
TGTCATTGCCACCATTGTCTCTTGTCACAATCCTAATTCTATGTTCTGATAATA
AAACTTCATTAGTGTCTGCACCTCCTTTCCCGTACCTTTGAAACACTGTCTTCT
GCTGAAACGATTAGCAGTGCGCTGCGTCAGATCATCCGGCTGTCTGACAGCCAT
TGTGAGTGAGCTAAATTGTGAGAAAATGAAAATTTCTGACAATTTCCATTATTG
TCAAATGGAGATATTTACTTGTACAAATATTAAAGGTTATTTAAAGATGTAAAC
GGACTTCATTTCATTAGTGGTATGCATCAGGTAATAATTTAAAACAACTACCCA
GCCTCATTCAAATAAATTACCAATT
>agCNE2
GAATAAATTATAGATGAAACCCCCATCATATGTTTATAAATACACTAACATTAG
CATTTTACTTCCTCATAAATCTGCAGCAAATACTCAGGCACAAGATAGAAAGCC
CTGTAAAACAATGATATATTCTCTGCTTGCAGGCATTGACTAGTGGCCCATTCA
CTCCCGGTGCTGTTCTTCAACCACGCTCCCTCCGCTCATGCCCCGTCATTTAAA
ATCCATTTTAATTTAAAGATGCACAAGGCAGTGCTCGTGGTACATTTACTCATC
CTAGTTATACATCATGGAGCAAGGAAAAGAAAATTGTTTCATCTAGAGATACGG
TTCAATGTGGTCACCACTGAACAGTGGAGTAATTACCATATCAGACCCACTCAA
AATGCACTCAGGGTGTTCTGTGACCTATCATTTGCTAATCTGCATAGTGAAGTA
TAGCTTCATAACTATAAATTGCTGACTTAATTAAAACCAAGACCTTTCTGTCAT
TTAATTACAGGGAGAAAGGTTATTGCTTAAATGCAATTTTCCAAATGTGATTTC
TTAACAGTTATTAACTGCTTCACCATCATTTATTGTTTCAGATTTTTCAATTTG
AAAATACTTCCCGTGACAGAAGTAAT
>agCNE3
TTACCCTGTGAAAGCCCCATCACTCACAGAAACTATTGCTGAGGAGGAGAGGAT
GATTTTATTTAGCTAATTTTTAACAAATCTCCAATGTTGCCCACATCTCCTGAA
GCTGGGAGGAGGGATAATCAAGTCAAGCCCCATTGACTAGGAGAATAGTGAACT
GTGGAA
|
| On you computer, open the dbCNS top pages using 3 browsing windows. Copy & paste these 3 CNS queries, agCNE1, 2, 3, to each (B) BLAST & alignment window, separately. Then push the SUBMIT buttons with mode (B). Analyses were confirmed to work for at least 10 simultaneous runs. |
 |
| |
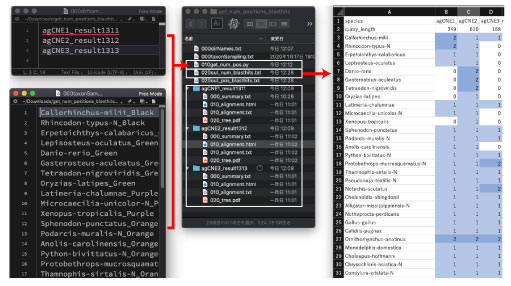
| Download results. Then change names of downloaded directories. These downloaded directories can be used in the subsequent section, Summarizing results. |
 |
| |
| |
| Summarizing results |
Using the downloaded pipeline, the number of blast hits can be counted from multiple results:
| 1. |
Download and decompress get_num_positions_blasthits.zip. |
| 2. |
From your terminal, cd to the get_num_positions_blasthits directory (see figure below). |
| 3. |
Run the pipeline: |
| |
python3 010get_num_pos.py |
| |
For your own data, copy and paste directories derived from "Multiple analyses (above section)" in the get_num_positions_blasthits directory. Then modify 000dirNames.txt and 000taxonSampling.txt files. |
| 4. |
Open the 020out_num_blasthits.txt file by your Excel. This result can be used to make a figure like Fig. 5A of Inoue and Saitou submitted. |
| 5. |
Coordinates of blast hits are saved in the 020out_pos_blasthits.txt file. This result can be used to make a figure like Fig. 5B. |
|
|
| |
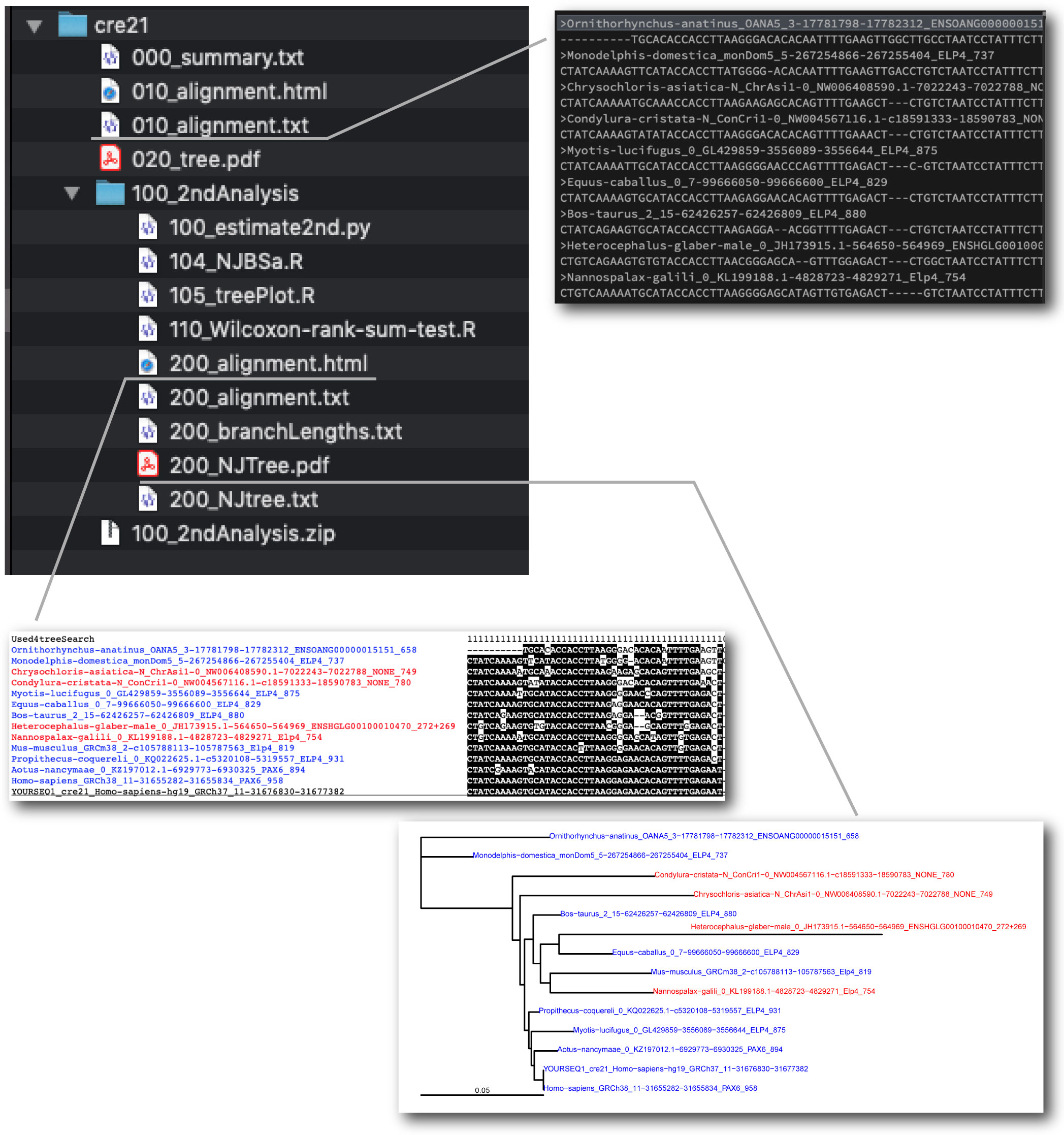
| Generating alignment and trees with focused CNSs |
|
By using selected sequences of dbCNS outputs, users can construct alignments and CNS trees on your computer. We made an analysis pipeline for this 2nd analysis. The script is specialized for a Macintosh use with Python 3. Windows users need some modifications.
Scripts:
100_2ndTree.zip
An example data: cre21.zip
|
Installing Dependencies
The 2nd analysis requires some dependencies to be installed in your computer.
MAFFT v7.407:
Available here: https://mafft.cbrc.jp/alignment/software/.
After compilation, set your PATH following this site.
TRIMAL v1.2 (Official release):
Available here: http://trimal.cgenomics.org/downloads.
cd to trimAl/source, type make. Then copy the executable.
make
cp trimal ~/bin
Ape in R:
R (3.5.2) is available from here.
By installing R, rscript will be installed automatically.
APE in R can be installed from the R console as follows:
install.packages("ape")
|
| The 2nd analysis |
The actual process is as follows:
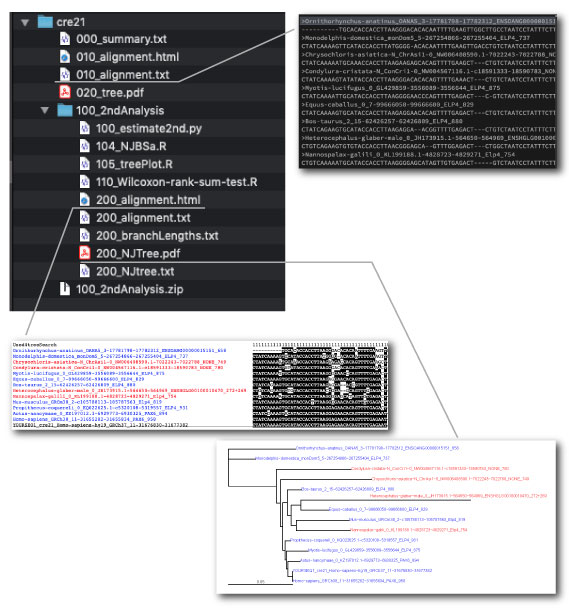
| 1. |
Decompress the cre21.zip file. |
| 2. |
Open the 010_alignment.txt file by your editor and delete extra sequences.
Based on the 1st alignment and CNS tree, users should select sequences manually.
Then save this file. |
| 3. |
From your terminal, cd to the 100_2ndAnalysis directory. |
| 4. |
Run the pipeline. |
| |
python3 100_estimate2nd.py |
| |
The pipeline align selected sequences using MAFFT (Kath et al. 2005). Multiple sequence alignments are trimmed by removing poorly aligned regions using TRIMAL 1.2 (Capella-Gutierrez et al. 2009) with the option “gappyout.” Phylogenetic analysis is performed with APE in R (Popescu et al. 2012) with the TN 93 (Tamura and Nei 1993) + gamma (Yang 1994) model. |
| 5. |
Automatically, the 2nd alignment is saved in the 200_alignment.html file and the 2nd CNS tree is saved in the 200_NJTree.pdf file. |
|
 |
|
| |